
当前新发突发病毒的传播对公共卫生已构成严重威胁,并可能在全世界引发毁灭性疫情。因此,加强对新发突发病毒病的检测和早期预警显得尤为重要。二代测序(next-generation sequencing, NGS)不仅能够精确地识别病原体,还可与生物信息学结合,在基于病原体基因组测序基础上,对新发突发传染病进行监测,进一步提升了临床诊断及微生物学认知水平。
来自佳木斯大学基础医学院和北京化工大学生命与科学技术学院的研究者们,综述了应用二代测序检测新发突发病毒病的病原体,并进一步研究其变异与进化特征、致病机制及传播规律,并将二代测序应用于临床疑难传染病的诊断,从而制定更有效的新发疫情防控策略。
中华实验和临床病毒学杂志:
DOI:10.3760/cma.j.cn112866-20220627-00144
21 世纪初,全球正面临全新的公共卫生问题,如高致病性的严重急性呼吸综合征冠状病毒(severeacute respiratory syndrome coronavirus, SARS-CoV) 和中东呼吸综合征冠状病毒(Middle East respiratorysyndrome coronavirus, MERS-CoV)[1-2] ,2013—2016 年非洲流行的埃博拉病毒(Ebola virus, EBOV)[3] ,以及现如今肆虐全球的新型冠状病毒(2019 novelcoronavirus, 2019-nCoV)[4] 。这反映了近年来由于自然环境或人为因素的改变,增加了新型病原体引起新发传染病在全球大规模暴发的可能性,同时,这也暴露了公共卫生系统在监测和预防新发突发病毒出现、传播等方面存在滞后性的问题[5-6] ,这需要研究人员加快对新出现的传染病进行诊断、持续监测和实时跟踪。然而,由于新出现的病毒适应性强且变异快,应用传统的分子生物学技术很难对其进行有效检测和预防。
二代测序(next-generation sequencing, NGS)可以无偏倚地在短时间内迅速筛查和识别新出现或正在发生变化的病原体信息,同时完成传统基因组学研究(测序和注释)以及功能基因组学研究(基因表达及调控、基因功能、蛋白 / 核酸相互作用),这为应对突发传染病事件、有效控制疫情、疫苗研发都起到了极大的促进作用[7-8] 。本文将讨论近年来 NGS 在新发突发病毒检测中的应用,并且应用该项技术进一步理解病原体的变异进化特征、致病机制及传播规律,这为 NGS 在临床上监测疫情发展、鉴定筛查疑难病原体等工作奠定了理论基础,并在新发传染病的防控工作中提供了重要的参考依据。
1
NGS 概述
NGS 平台
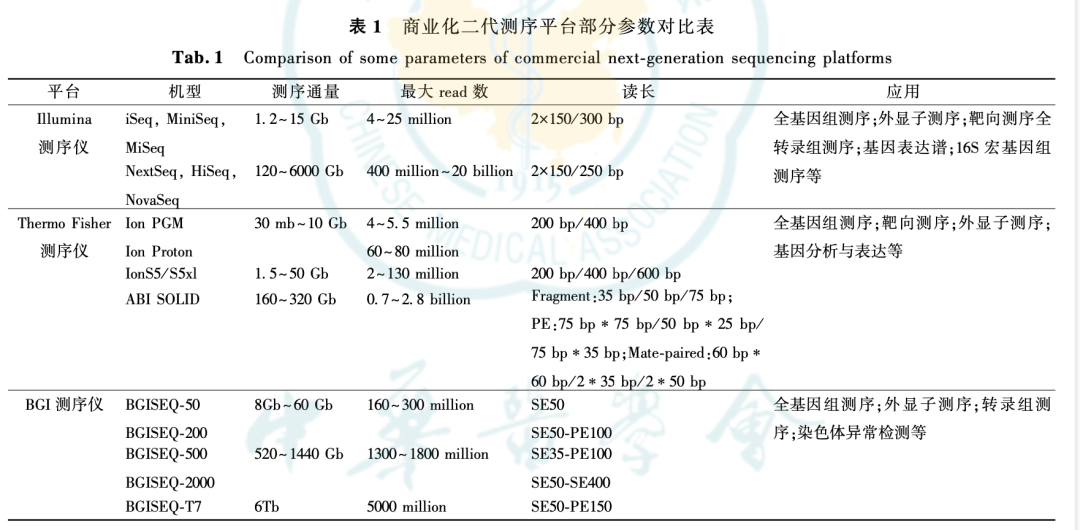
NGS 属于高通量测序 (highthroughput sequencing, HTS)[9] ,HTS 主要有两种工作模式,分别为短读长测序和长读长测序,都能够同时对数亿条 DNA 片段进行测序[10] 。二代测序一般是短读长测序,主要是提供成本更低准确性更高与临床变异相关的数据,长读长一般是指三代测序,也叫单分子测序,主要用于解决大范围的复杂的基因组学问题,例如复杂基因组组装、单体分型、复杂结构变异和基因同种型鉴定[11] 。目前商业化 NGS 平台主要有 Illumina 公司提供的 MiniSeq、MiSeq 等为代表的小规模测序仪以及 NovaSeq 为代表的大规模测序仪;Thermo Fisher 公司提供了 Ion Torrent 系列;华大基因提供的 BGISEQ 平台以及罗氏公司 454 焦磷酸测 序 454 GS Junior + 和 454 GS FLX TitaniumXL +(罗氏测序仪现均已停产)[12-13] 。不同的测序平台根据不同的实验需求,选取适合序列读长的测序试剂盒,来满足实验的要求(见表 1)。目前 HTS仪器的出现,已经将微生物全基因组测序从价值百万美元的科研活动转变为分子生物学的常规操作[14]。
二代测序优势
传统的临床微生物学检测病原体技术主要有微生物的培养与分离、病原体特异性抗原、抗体(血清学)检测、生化免疫指标及通过聚合酶链式反应(polymerase chain reaction, PCR)来鉴定微生物的核酸分子(DNA 与 RNA)[15] (见表2)。高灵敏度检测对于临床检测病原体和公共卫生研究至关重要。以 PCR 等诸多基于核酸检测的方法为例,其在单个病毒检测中因具有高灵敏度(PCR 方法可检测 1~10 拷贝/ml 的病毒载量)[16]和低成本的优势,被广泛应用于检测病原体。然而,常规检测方法受到多种因素的限制:首先需要提前了解所靶向的病毒,以及通常在单个测试中所需靶标数量。这使得在面对新发突发病毒时,由于没有可以参考的病毒基因组信息,这些检测方法无法有效开展。其次,由于病毒基因组进化而发生变异,尤其是大部分 RNA 病毒在复制过程中缺乏校对机制,频繁出现基因突变,导致在特异性 PCR 检测时不能有效识别靶病毒。这对病原体的精准鉴定带来许多障碍[17] 。NGS 的出现启动了宏基因组学领域的研究,不需要微生物培养以及靶向病原体序列信息就可以无偏倚地筛查某些地理区域中所潜在的新型病原体( 病毒、细菌、真菌和寄生虫) 。

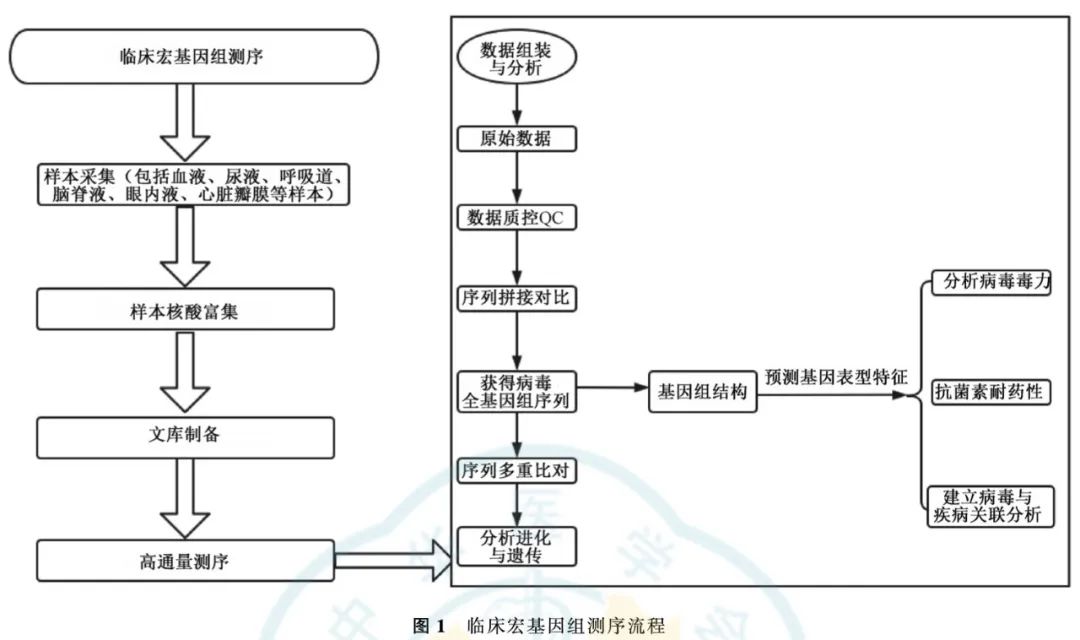
测序基本分析流程
临床宏基因组学主要检测步骤包括:(1) 标本前期处理;(2) 临床样本中核酸富集,相关文库构建,进行下一代测序;(3) 结合生物信息学方法处理原始数据;(4)组装的基因组数据用于预测表型特征,例如病毒毒力、病毒的进化特征、抗菌素耐药性和血清型等;(5)基于全基因组序列进行进化与溯源分析,从基因层面对病原体进行进一步的遗传表征分析;(6)数据上传数据库并公开,供其他研究人员进一步用于诊断、治疗和疫苗的开发[18-19] 。
NGS 与生物信息学相结合,已成功应用于传染病研究中。目前已开发了各种计算工具用于分析测序后的大量数据,高效处理这些数据是表征各种病原体的关键。生物信息学需要足够的基础设施,包括网络的存储系统、分析通道、完整的湿实验流程、测序平台,以及专业人员。测序结果反馈流程首先对来自测序平台的原始数据去除宿主基因组、清理修剪和过滤以消除低质量和重复的序列。其余的序列通常是进行组装,将来自同一物种的序列整合到更长的拼接序列中。一些常用的组装的工具包括Megahit、SOAPdenovo、 Velvet 等。而 后 将 获 得 的 重叠群与核苷酸或蛋白质数据库中的序列和基因组进行匹配,并对所得重叠群进行分类鉴定,如 Kraken、Kraken HLL、MEGAN6/EC,BLAST 通常是最常用的工具。一旦组装出高质量的基因组,就可以进行其他类型的分析,如重构病原体的传播链以及跟踪耐弊端价格较昂贵;实验较复杂;需要除去人类宿主背景进行测序;基因组测序结果易被环境物种污染价格较昂贵;测序步骤更复杂;基因组测序结果易被环境物种污染周期长;某些微生物不能在现有的培养技术下生长敏感性低;特异性低易受到特异性或得特异性抗体影响出现假阴性假阳性结果;免疫功能异常影响检测结果存在一些假阴性、假阳性等问题需要特异的引物;仅限于基因组的一小部分;模板拷贝数较低时,检测的精确性低很难开发和用于检测具有高表位突变率的病原体;需要昂贵的实验室设备药性的选择,从而有助于流行病学调查[20-21] 。临床宏基因组学和全基因组测序以及其他新兴NGS 诊断技术,克服了传统病原体检测存在的局限性,成为更精确表征和鉴定病原体特征、研究其遗传变异以及后续抗病毒药物研发的最佳工具,未来将广泛应用于临床诊断和治疗(见图 1)。
2
NGS 在病毒病检测中的应用
发现和鉴定新型病原体
据估计,目前有 160万种以上尚未被发现的病毒种类存在于哺乳动物和鸟类宿主中[22] ,这些病毒很有可能存在人畜共患感染的风险。NGS 作为一项不依赖于微生物培养分离的新型检测技术,可从难以诊断的病例或各类病原体宿主中发现潜在的致病病原体。例如,继 2009年刚果民主共和国暴发致命性急性出血热疫情后,Grard 等[23] 使用 NGS 测序组装和表征急性患者血清样本中新型横纹病毒(Bas-Congo 病毒或 BASV)的基因组。2009 年,在中国中部和东北部地区,多个科研团队应用 NGS 确定了一种由蜱虫传播的严重发热伴血小板减少综合征病毒(severe fever withthrombocytopenia syndrome virus, SFTSV),感染 后其临床表现为重度急性发热并有严重白细胞、血小板降低,感染初期死亡率可达 30% [24-25] 。
在新型冠状病毒肺炎 (Coronavirus Disease2019, COVID-19)疫情期间,NGS 同样也发挥了至关重要的作用。Zhou 等[26] 基于 NGS 平台从多名患者样本中获得 2019-nCoV 全基因组序列,快速鉴定出引起此次疫情的病毒与 SARS-CoV 具有79. 6%的同源性,与 MERS-CoV 同源性为 50%左右,同属于β冠状病毒。根据其他研究团队通过 NGS 获取的序列分析表明,2019-nCoV 与 2013 年检测到的蝙蝠冠状病毒 RaTG13 序列同源性为 96%,与从马来西亚的穿山甲中分离出的冠状病毒 GX_P2V 基因组具有85.5%~92.4%的同源性[27-28],这提示部分哺乳动物可能作为 2019-nCoV 的传播宿主,存在人畜共患病的风险。综上可见,在面对新发突发疫情时,使用NGS 快速鉴定和表征潜在致病因子,可为后续追溯病毒起源和分析病毒进化特征,以及抗病毒药物和疫苗研发工作打下了坚实基础。
研究病毒基因组变异与进化特征
大部分RNA 病毒在合成过程中由于缺乏校对功能,或存在复制过程相对不稳定性导致病毒突变频繁发生,这种遗传变异将影响病毒的某些表征,如趋向性、毒性以及对现有疫苗和抗病毒药物的敏感性,还可能造成疫情大流行。因此,研究这些变异及其相应的突变对疫情发展趋势和疫苗研发至关重要。
2019-nCoV 是使用 NGS 有效应对新发传染病大流行的典型案例,2019-nCoV 通过刺突(spike, S)蛋白与人血管紧张素转化酶 2(angiotensin convertingenzyme 2, ACE2)结合,因而大多数单克隆抗体和疫苗将S蛋白作为靶点,而病毒在这一区域的突变可能影响现有单克隆抗体和疫苗的效力[29] 。NGS 为病毒变异分类提供依据,首先用于描绘英国报道的B.1.1.7(Alpha)变异体,其 S 蛋白突变主要包括N501Y、P681H 以及氨基酸 69 / 70 位点缺失,虽然这些突变尚未影响疾病发病的严重程度或疫苗的效力,但初步流行病学调查表明,这些突变与病毒传播性增加密切相关[30]。随后B.1.351(Beta)谱系于南非被发现,并在该地区迅速传播,掀起该地区的第二波疫情浪潮,P. 1(Gamma)谱系于巴西确定,相较于 Alpha,Beta 和 Gamma 的感染性相对较弱,但它们具有较强的免疫逃逸能力,这是由于 E484 干扰了中和抗体与对应表位的结合,使得疫苗接种者的血清对 B. 1. 351 和 P. 1 突变株的中和活性受到影响,出现突破性感染几率增加[31-32] 。B. 1. 617. 2(Delta)谱系于印度发现,是继 Alpha 之后,具有与以往关切变异株(variant of concern, VOC) 不同突变的病毒株,在病例样本测序中,Delta 占据了主导地位,已成为全球 2019-nCoV 流行的优势毒株。这是由于L452R、P681R 的结合增强对细胞的感染,并且L452R 和 T478K 均能增加 Delta 的免疫逃逸能力[33-34] 。2021 年 11 月,一种名为 B. 1. 1. 529(Omicron)的新 VOC迅速于非洲出现,研究小组对所获得的临床样本进行靶向全基因组测序。其 S 蛋白上的突变高达 30个以上,使得该谱系可能具有更广泛的传播性以及对当前 COVID-19 疫苗免疫具有更高的耐受性,并成为世界主要流行的 VOC[ 35-36] 。2019-nCoV 基因组任何的变异都可能造成严重的公共卫生后果,这些新型毒株的出现都需要在全球的临床实验室中对2019-nCoV 基因组进行测序,对这些变异株进一步分析将有助于理解 2019-nCoV 感染人类的相关机制,进而在新发突发疫情时调整相应防治策略。
自 2014 年初至 2016 年末,EBOV 在非洲广泛传播期间,其极强的致病性引发各界忧虑,各国研究组通过 NGS 提供了大量的 EBOV 全长基因组序列,以更好地了解当前疫情的病毒进化和传播动态。Schibler 等[37] 对 1 名在塞拉利昂感染 EBOV 的患者进行测序,结果表明 EBOV 基因组处于稳定的状态,只有 5 个同义核苷酸的变化(4901A→C、7837C→T、8712A→G、9947T→C、16201T→C)。EBOV 基因组复制过程中容易发生错误,1 项测序研究显示EBOV蛋白 VP30、 VP40 和 VP24 的变化范围很小,而在GP、VP35、NP 和 L 中超过15%的病毒基因组中存在某些氨基酸变异[38-39] 。通过 NGS 发现,在 2014—2015 年暴发的西非 EBOV 疫情中,病毒的突变率与以前的暴发突变率相比没有显着差异,碱基替换速率较为平缓,总体变异水平仍然很低,没有在人类中经历快速进化[40] 。因此,NGS 可在较短时间内快速地对整个病毒基因组进行测序,以便了解病毒的基因组特征进而监测病毒变异与进化情况。
研究病毒致病机制
新发突发病毒通常明显区别于以往的病毒,获得其基因组序列了解其发病机制对疫情防控工作尤为重要。目前导致 2019-nCoV 流行的潜在致病机制仍然是悬而未决的问题,Wang 等[41] 使用 NGS 在感染 2019-nCoV 后的多个时间点对不同细胞系进行测序,鉴定了数百个模板开关并构建了 2019-nCoV 亚基因组的演化动态,进一步研究了这些模板开关连接位点之间的配对规律,发现了 RNA-RNA 相互作用的基本模式,研究结果揭示了 2019-nCoV 的亚基因组演化动态和调控特征,为研究其发病机制和开发新型抗病毒策略奠定分子基础。Rueca 等[42] 应用 NGS 对感染 2019-nCoV 患者的上呼吸道和下呼吸道分泌物进行全基因组和宿主内变异性特征的研究,研究结果观察到患者体内变异相当低并且沿病毒基因组均匀分布,表现出遗传异质性和准种区室化。此项监测 2019-nCoV 基因组特征的研究更好地了解宿主与病原体之间的相互作用,为分析 2019-nCoV 致病机制提供了新视角[42] 。
最近,Wang 等[43] 利用严重发热伴血小板减少综合征(severe fever with thrombocytopenia syndrome,SFTS)患者的白细胞进行 RNA-seq 和 RNA 甲基化免疫沉淀测序(m6A-seq),分析了此病毒的转录组学和表观遗传学特征,发现 SFTSV 可能诱导死亡的相关因素(血小板急剧减少和先天免疫应答延迟),并根据感染期基因表达变化推测基因调控途径。此项测序和临床相结合研究对了解 SFTSV 致病机制具有重要意义,同时有益于监测 SFTS 疾病的进展并提供临床治疗指导建议。
描绘病原体传播规律
基因组测序数据丰富了全球数据库并对研究流行病学特征具有指导意义。通常当病毒基因组测序时,需要对 NGS 文库进行靶向富集,如使用多重 PCR、捕获探针、具有加标引物富集的宏基因组测序等文库富集策略以获取足够的病毒基因组覆盖率,进行系统发育和分子钟分析,并且已成功应用于 2019-nCoV、EBOV 的基因组监测,以及包括与载体传播(如虫媒病毒)相关的发热性疾病和出血热疫情中[8] 。如在非洲暴发 EBOV疫情时期测序显示,所有毒株均具有相关性,病毒可发生性传播[44] 。而后多个研究小组从基因序列分析描述了流行于西非的 EBOV 毒株的传播特点,多个测序样本分析表明引起本次疫情的 EBOV 毒株与早期 EBOV 单个病毒谱系传播模式不同,出现了多个病毒谱系(A、SL2、SL3)共同传播,在人际之间传播增强,这使得该病毒存在大量非同义突变和同义突变的积累[45-46] 。寨卡病毒(Zika virus, ZIKV)测序能够确认该病毒在美洲的起源和传播情况,该病毒起源于巴西,并通过美洲传播[47] 。测序数据揭示了多数本地 COVID-19 病例是由其他病毒活跃性地区的输入造成的,并非来自本地传播链大范围的传播。随着国际实施大规模基因组测序以及采取封控管制措施,序列多样性逐渐减少,NGS 有效地干预了病毒在人口密集地区的传播链,基于基因组测序方法将帮助研究人员监测病毒的传播和进化动态,对于预防病毒在未知地区传播具有重要意义[48] 。
应用于药物和疫苗的研发
应用 NGS 识别复杂样本中的单个病毒基因组,检测小变异体,提供了实时的毒株序列信息以监测疫情和研发特效疫苗、药物。例如,COVID-19 大流行期间,用于 NGS 的商业试剂盒(Illumina Inc. , USA)已被批准用于临床诊断检测,NovaSeq 测序平台可同时运行高达 3 000多个样本,差异诊断设计包括 98 个扩增子,并在 24h 内完成病毒 RNA 提取、文库测序以及提供准确、高灵敏度结果,并且其检测结果与 RT-PCR 具有高度一致性[49-50] 。当前潜在高传染性和高致病性的新 VOC 接连出现,迫切需要开发简便和具有低成本的方法,以便在社区、医疗机构等每周对数千个病毒样本进行测序。Simonetti 等[51] 提出了一个更简化的工作流程 COVseq,可构建与 Illumina 平台兼容的测序文库,明显降低了文库制备和测序成本。COVseq 可立即适用于 2019-nCoV 基因组大规模监测,并且其他病毒也同样适用,如流感病毒。当2019-nCoV基因序列公开共享并上传到 GISAID、GenBank 等数据库时,在几周内对整个 2019-nCoV基因组进行了测序,为了解和跟踪感染动态、研究2019-nCoV 新 VOC 特征提供帮助,使得在 6 个月内开发基于 mRNA/DNA 平台的有效疫苗成为可能[52] 。在治疗疾病之前,NGS 可以对预先存在的耐药突变体进行基因筛查,对于特定药物进行遗传筛查,以便检测临床病毒分离株中的耐药性突变[53] 。
NGS 在获取病毒基因组序列预测药物靶点时也发挥了积极的作用,有研究表明通过测序识别乙型肝炎病毒(hepatitis B virus, HBV)和丙型肝炎病毒(hepatitis C virus, HCV)基因组序列中高度保守的区域,可能会揭示基因的保守区域和治疗的靶点[54]。在由HBV所致肝细胞癌(hepatocellularcarcinoma, HCC) 的治疗过程中,基因治疗似乎是治疗 HCC 或降低 HCC 风险有希望的策略之一[55] 。临床应用 NGS 展示了来自 EBOV 感染患者的总计710个宿主内单核苷酸变异 (intra-host singlenucleotide variation, iSNV) 的全面分布情况,iSNV 及其等位基因频率分析表明 VP40 属于保守基因。最新研究表明合适的核酸可以触发 VP40 从二聚体动态转化为八聚体环状结构,VP40 环可以结合宿主细胞 mRNA 的 3′ UTR 中的结构序列。这种结构转变是病毒生命周期必不可少的一部分,因此 VP40 可能会成为设计有效抗病毒药物的靶点[56-57] 。
3
总结与展望
NGS 在临床微生物学研究中作为一种诊断各种新发突发传染病的有效工具,其不依赖于特定的引物和探针就能鉴定未知病毒,同时能够及早发现病原体传染性风险,在疫情大流行初期及时采取监管措施,从而防止高度传染性病毒自由传播。特别是在现阶段 COVID-19 疫情及其他流行病传播背景下,NGS 能够检测并恢复与血液传播感染相关的各种新兴病毒基因组,这对于了解病毒传播趋势和动态,研究病毒的生命周期,调查潜在的流行病学和疫情来源,协助开发有效疫苗和抗病毒药物具有重大意义。
目前尽管 NGS 在新发突发传染病的研究中具有突破性意义,但将其大规模应用于临床常规检测仍存在以下方面的挑战。第一,NGS 在序列读取方面长度有限,可能会导致基因组序列错误组装或者基因组覆盖不全,造成读取序列与同源区域比对结果不匹配,可能会出现假阴性或假阳性的结果,现已经创建了许多工具用以注释具有高度同源性的已知序列区域来解决此问题(如 https://www.ncbi. nlm.nih.gov/books/NBK535152/以及其他正在开发的工具用于强制对齐到感兴趣的基因组区域,并帮助纠正未对准的测序区域)[22,58] 。第二,短读长 NGS 平台的优势是精确发现小变异体,如单核苷酸变异体(single nucleotide variation, SNV)、插入 / 缺失(insertionand deletion, indel)[59] ,这些变异可能会影响某些病毒表型特征,但对于大的结构变异(structural variation,SV),使用短读长测序无法确定其等位基因特异性表达位点,这是一个重要的问题,因为 SV 与许多严重的疾病密切相关[60-61] ,因此 NGS 短读长平台不适用于检测复杂基因组位点或者不规则的复合结构,但可以应用长读长三代测序技术检测复杂区域的SV[62-63] 。第三,如今 NGS 个人全基因组测序成本已经从数十万甚至数百万美元下降至 600 美元 / 人,但相比于其他常规检测仍然成本较高。为此一方面需要确保该检测技术的临床有效性,即在针对某些特定适应症情况下,该技术具有最高的应用潜力。生物信息学中心也应尽可能降低数据分析成本,以提高这些测定在临床中的可用性。另一方面公众或私人支付者需要足够的报销水平,做出有利的保险决策以支持 NGS 的应用与发展[64] 。
随着 NGS 临床应用的突飞猛进,在实际应用过程中难免因为检测技术的门槛低、缺乏临床公认的指导规范等原因,导致疾病诊治中不可避免地出现检测质量差、结果分析不够专业等问题,因此必须促进标准化。目前,新发突发传染病广泛流行,这使得对基因检测的需求急剧攀升,国内外开发多种 NGS试剂盒应用于疾病诊断。应当注意的是,一方面,新发疫情背景下的需要调查的数据往往非常广泛,在海量的数据中找出关键致病病毒并非易事。另一方面,各检测机构也拥有了大量的个人基因组测序信息。如何确保有效利用数据以及整合海量的测序数据,建立标准化的数据库[65] 和制定实施规则均是NGS 未来标准化发展的关键要素。因此监管部门应尽快审查和制定各种标准化的规范与指南,以确保 NGS 行业有序快速的发展。
NGS 作为一项综合性的直接检测方法,目前较高的成本使其短期内无法成为临床一线检测方法,但它仍有潜力成为困难和复杂疾病、免疫缺陷及其他特殊人群中病原体鉴定的准一线检测方法。与任何新技术一样,NGS 也需要严格的临床实践考验。为了保证所得测序结果的准确性,需要对测序平台灵敏程度、实验中每一个测序环节进行质量控制,并且测序所得结果不能作为临床诊断唯一标准,需要与临床实践和基础理论知识相结合。
NGS 作为一项具有突破性的新兴技术,具有其他常规病原体检测无法比拟的优势,相信其在未来新发突发传染病病原体的发现、检测、监测和变异跟踪等方面能发挥更为重要的作用,提升研究人员对病原体的了解,为决策部门制定更有效的公共卫生干预策略提供及时的数据支撑,为传染病疫情的防控工作提供强大的技术支持。
北京微未来科技有限公司
北京微未来专注于病原微生物领域的新一代基因测序完整解决方案,研发和转化了一系列具有广泛应用前景的病原微生物基因捕获试剂盒、基因测序技术和生物信息分析平台。
解码病原微生物,致力传染病防控
MicroFuture,for Better Health.
公司地址:
北京市昌平区回龙观镇龙域中街
1号院1号楼B座704、605
办公电话:
010-62998049
售后电话:
400-666-0670